
 Force Field X
Force Field XWelcome to Force Field X
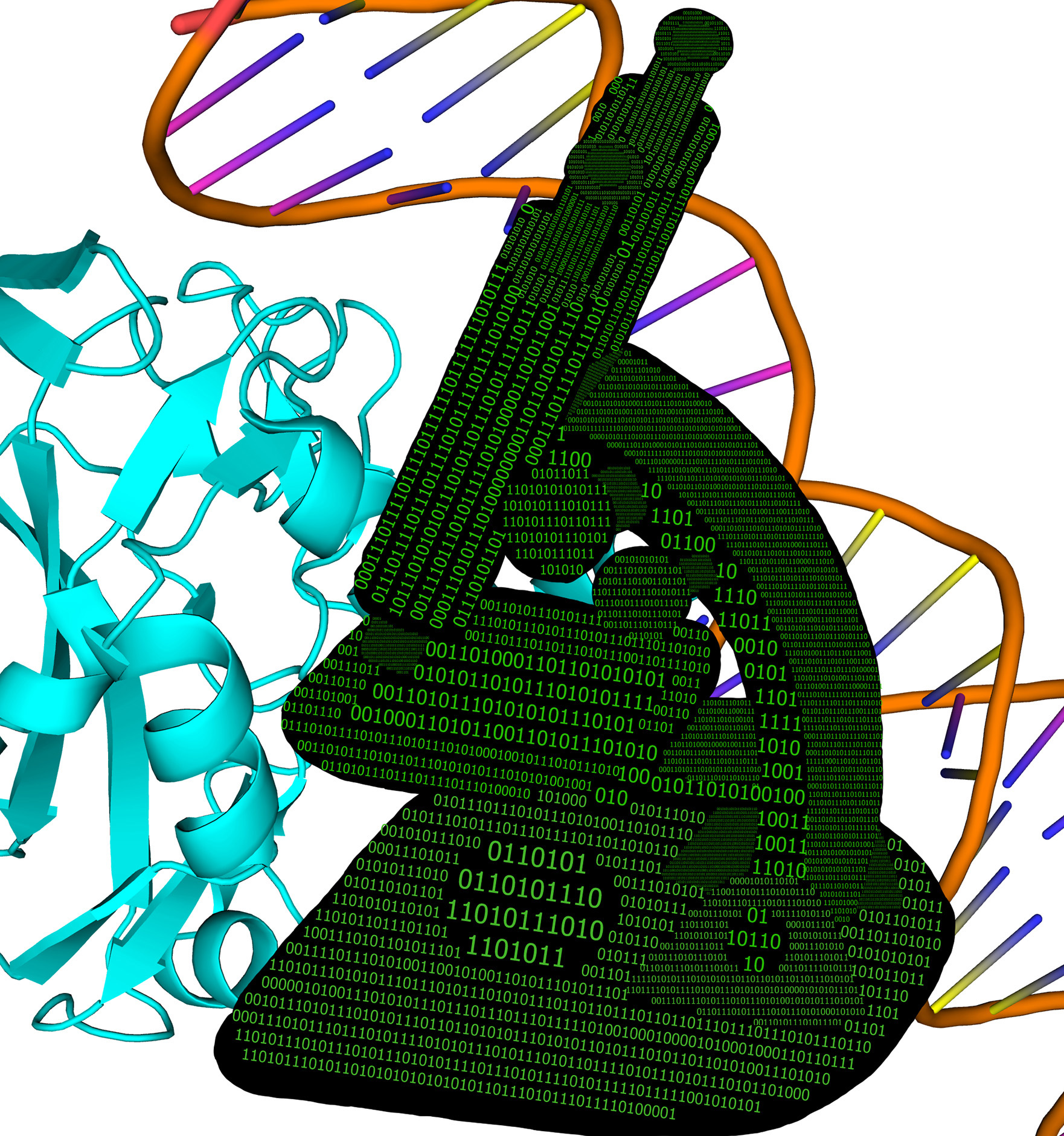
A Computational Microscope to Study Genetic Variation and Organic Crystals Using Theory and Experiment
|
The FFX platform targets open research questions in the areas of:
Performance
|
|
Getting Started
Instructions on how to download, install and use Force Field X are available in the following sections.
|
Download
|
Install
|
User Manual
|
Applications
|
|
Method Development
|
Build from Source
|
Programmer Guide
|
Publications
|







Funding Support
Development of Force Field X has been supported by the following organizations.
|
|
|
|
|
|
| NSF CHE-1751688 | NIH R01 DC012049 | SFARI Award 1019623 | U. of Iowa Mid-Career Scholar Award | Butch Pedersen Legacy Foundation |
| NSF CHE-2504153 | NIH T32 GM152268 |




